Программный пакет молекулярного моделирования Hess
Основой программного пакета Hess являются реализации алгоритмов вычислительной органической химии. Пакет позволяет решать ряд прикладных задач; основным применением Hess является виртуальный скрининг баз данных органических молекул путём оценивания энергии связи биоактивной молекулы (лиганда) с активным центром белка, входящим в изучаемый сигнальный путь.Функции
Подготовка данных
- Загрузка и сохранение файлов в форматах MOL/SDF, PDB, PDBQT, MOPCRT;
- Ароматизация, деароматизация;
- Восстановление структуры связей по координатам атомных ядер;
- Восстановление водородов;
- Протонирование/депротонирование;
- Анализ структуры молекулы, удаление ионов;
- Выделение части структуры молекулы (фрагмента).
Расчёты
- Вычисление полной энергии молекулы или комплекса молекул полуэмпирическим методом (PM7);
- Учёт водной среды с помощью неявной модели (COSMO);
- Вычисление полной энергии молекулы или комплекса молекул методом силового поля (Amber, CHARMM);
- Вычисление полной энергии молекулы или комплекса молекул гибридным методом SE/MM с учётом разорванных связей методом «Link atoms»;
- Вычисление энергии связывания лиганда и белка эмпирическим методом (Vinardo);
- Ведётся работа по вычислению энтропийного вклада в свободную энергию методом Монте-Карло.
Оптимизация
- Глобальный перебор положений и конформаций молекулы: метод Монте-Карло;
- Локальная оптимизация геометрии: метод BFGS.
Программные и аппаратные средства
Функции пакета Hess доступны как через программные интерфейсы (API) на языках C, C++ и Python, так и через приложения командной строки, осуществляющие подготовку молекул, расчёты энергии связывания (с полной оптимизацией, локальной оптимизацией или без оптимизации) и сохранение результатов.
В настоящее время Hess работает на рабочих станциях и серверах с CPU x86 и ARM; поддерживается параллельное выполнение на многоядерных процессорах. Ведётся работа по адаптации Hess к высокопроизводительным кластерам (HPC) и графическим процессорным ускорителям (GPU).
Код
Hess написан на языке C++ и имеет зависимости в виде отдельных модулей из следущих пакетов:
Примеры расчётов
Расчёт энергии связывания при наличии начального приближения
Рассмотрим пару лиганд-белок в комплексе 1WUM (NP2-PCAF) из базы данных PDB. Связывание лиганда (ацетилированного лизина) в бромодомене PCAF является частью процесса трансактивации Tat, одного из белков ВИЧ. Положения атомных ядер в данном комплексе вычислены на основании наблюдения ядерно-магнитного резонанса (NMR), т. е. близки к реальным положениям в растворе, в отличие от большинства других комплексов в базе данных, наблюдённых в кристаллической решётке.

Анализ PDB-файла показал, что в терминальном остатке белка — лизине — не хватает двух кислородов, формирующих замыкающую цепь COOH-группу; видимо, при ЯМР-спектроскопии эти кислороды были идентифицированы как кислороды воды.
После добавления потерянных кислородов требуется восстановить в структуре водороды. Изначально присутствующие в PDB-файле водороды удаляются, после чего на основании положений атомных ядер вычисляются кратности связей, включая ароматические; затем производится деароматизация и вычисляются формальные заряды и количество и положения водородов при тяжёлых атомах. При вычислении формальных зарядов и количества водородов учитывается влияние раствора (pH = 7.4).

Аналогичная подготовка производится с лигандом.

Общий заряд лиганда +1, общий заряд белка +5, следовательно заряд комлекса +6. Эти величины важны для последующего вычисления самосогласованного поля (SCF).
Рассчитаем энергию связывания по формуле Ebind = ENP2+PCAF − ENP2 − EPCAF. (Каждый член в правой части выражения является полной энергией или теплотой образования соответствующей молекулы или комплекса.) Расчёт энергии осуществляется полуэмпирическим (SE) методом PM7, при этом самосогласованное поле вычисляется методом локализованных молекулярных орбиталей (MOZYME) с использованием неявной модели водной среды COSMO. Каждый из трёх показателей рассчитывается с оптимизацией положений атомных ядер методом BFGS.

| ENP2+PCAF | = | -179009.490 эВ |
| EPCAF | = | -176569.552 эВ |
| ENP2 | = | -2437.847 эВ |
| Ebind | = | -2.091 эВ |
Вывод: подтверждено связывание лиганда с активным центром белка с энергией 2.091 эВ = 48.22 ккал/моль.
Расчёт энергии связывания при отсутствии начального приближения
База данных DUD-E содержит сведения о лигандах с экспериментально подтверждённым связыванием для 102 белков. В частности, для белка NRAM известно 98 таких лигандов. При этом ни энергии связывания, ни взаимных положений атомных ядер в базе данных не содержится. Проведём молекулярный докинг каждого из 98 лигандов в белке NRAM.
Прежде всего вычислим положения атомных ядер белка в растворе с помощью оптимизации энергии, используя координаты из DUD-E (рассчитанные методом кристаллографии) как начальное приближение. При расчёте поместим в активный центр один из лигандов. Произведём два варианта оптимизации: с оценкой энергии методом PM7 и методом молекулярной механики (MM) с использованием модели силового поля Amber.
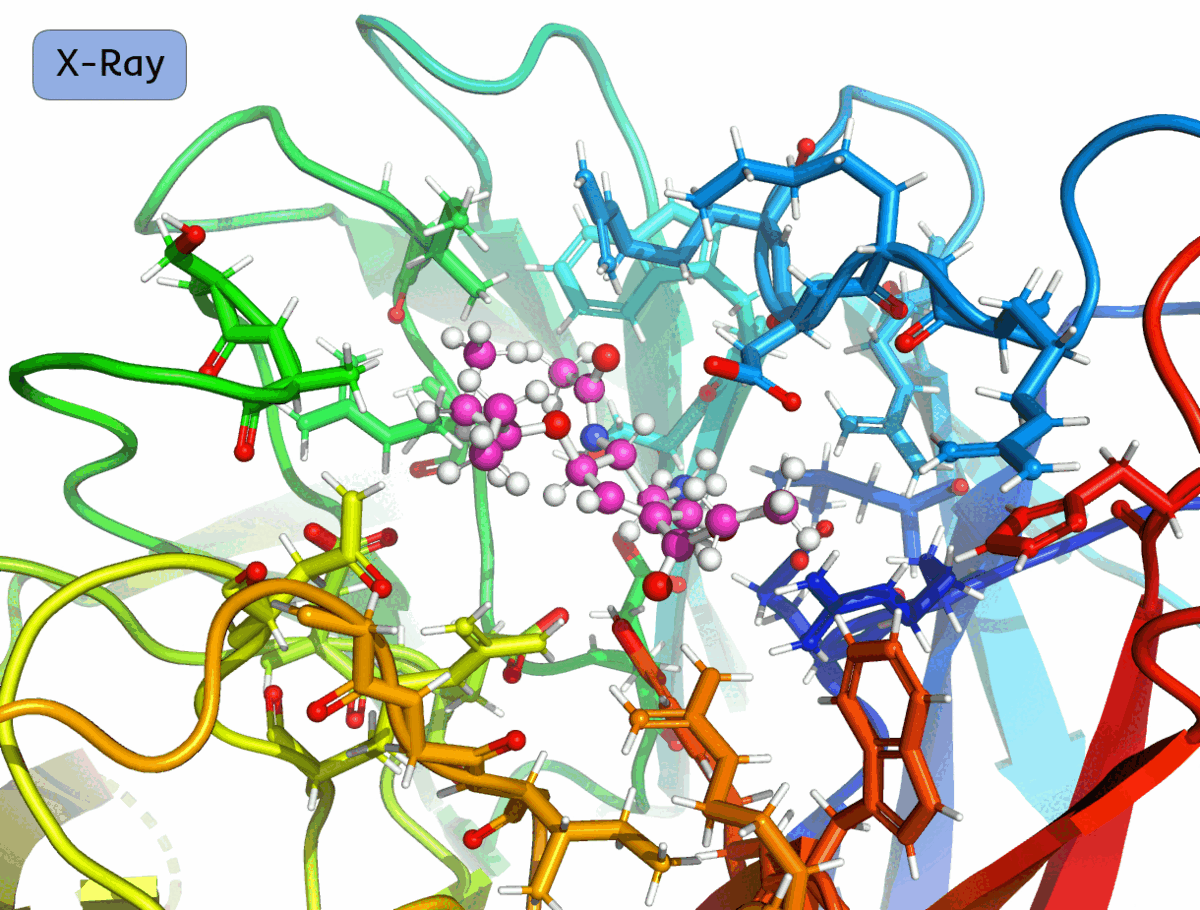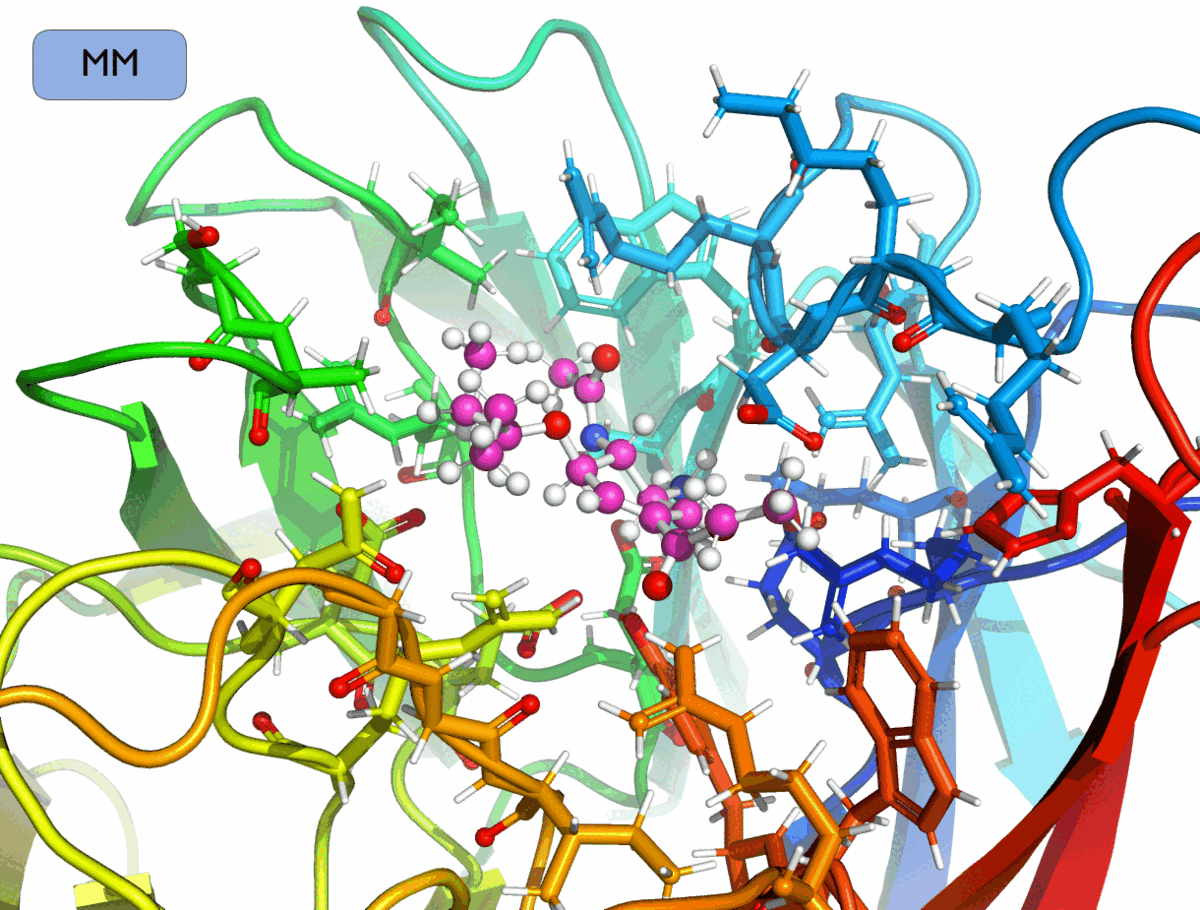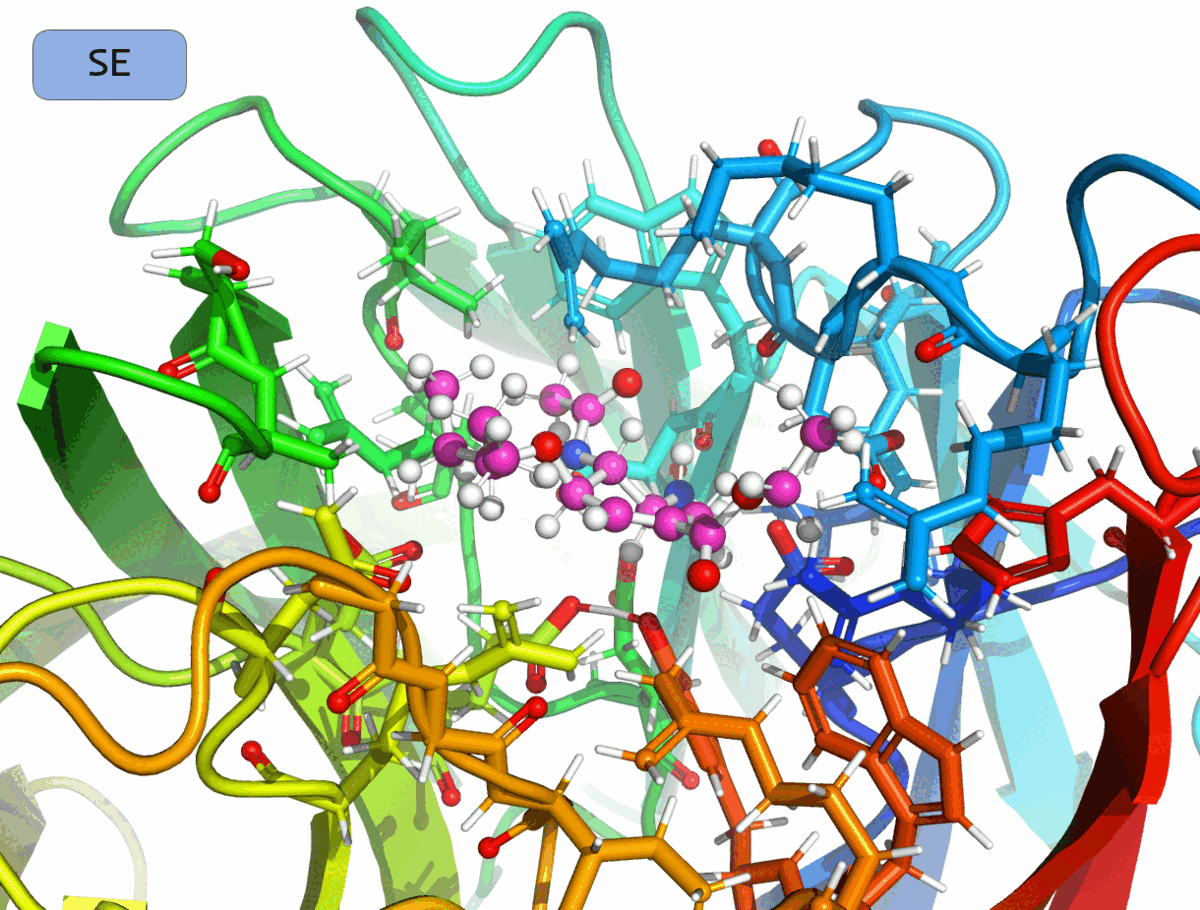
Для каждого лиганда требуется произвести глобальный поиск положения и конформации в активном центре NRAM. (Будем называть совокупность положения и конформации лиганда позой.) Поскольку такая процедура требует многократного оценивания энергии связывания для различных поз, на этом этапе воспользуемся эмпирической оценочной функцией Vinardo и применим метод глобального поиска Монте-Карло в (N + 6)-мерном пространстве, где N — количество вращаемых связей лиганда.
Проведём глобальный поиск оптимальных поз для всех лигандов, зафиксировав положение атомных ядер белка, полученные по результатам оптимизации по модели Amber.
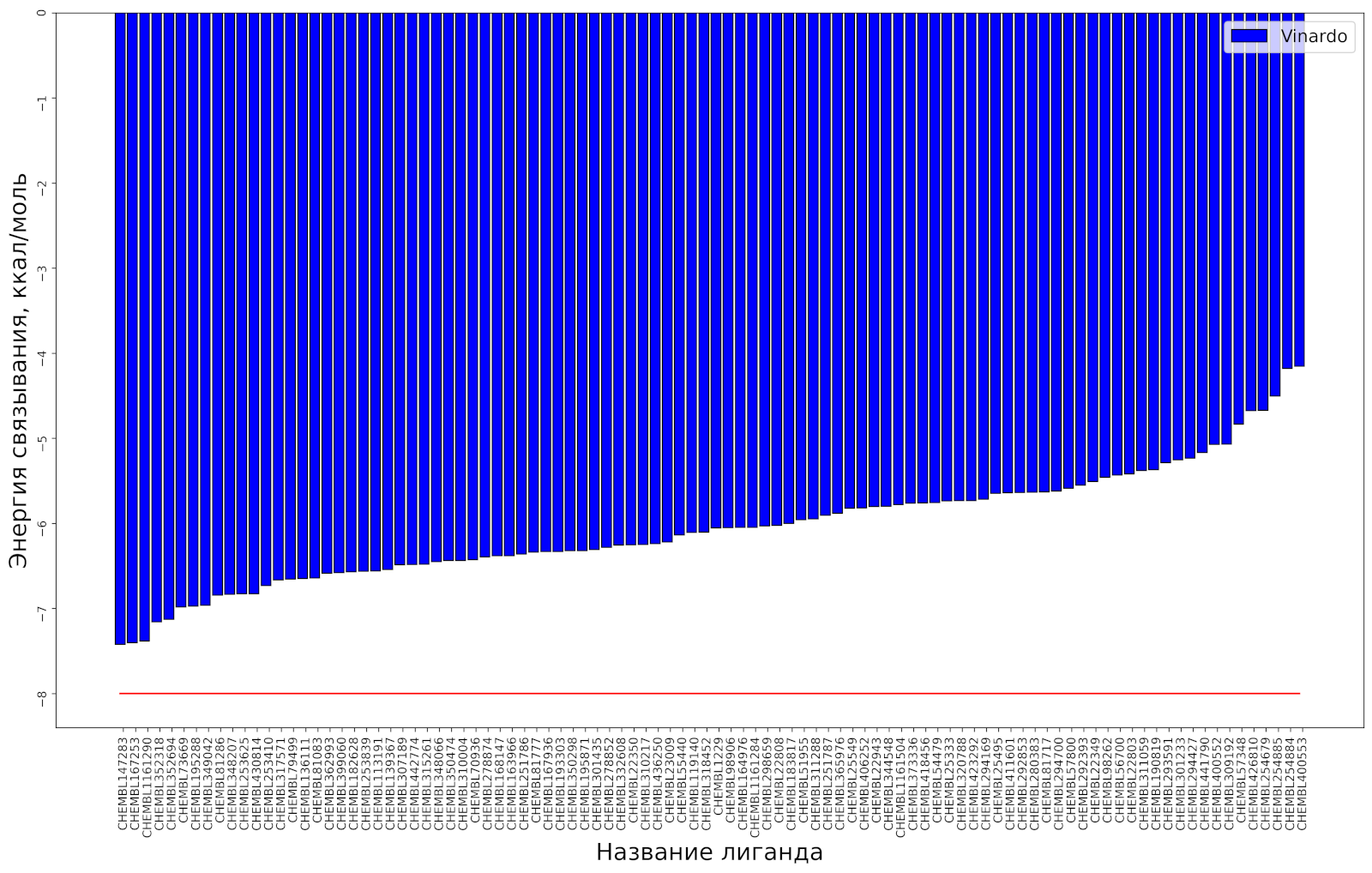
Формально для всех 98 лигандов нашлись позы, в которых оценочная функция принимает отрицательное значение, т. е. присутствует связывание; однако во всех случаях оценка не превышает барьер 8 ккал/моль, т. е. не является показателем устойчивого связывания для лигандов, содержащих 10 и более тяжёлых атомов (1, 2). Но полученные позы можно использовать как начальные приближения для расчёта энергии связывания более точными методами.
Исследуем адекватность гибридной (SE/MM) оценочной функции, более сложной, чем эмпирическая Vinardo, но более простой по сравнению с полуэмирической PM7. Выделим в белке фрагмент из аминокислот, близкий к активному центру, который вместе с лигандом будет отнесён к полуэмпирическому (SE) расчёту, в то время как оставшиеся аминокислоты будут отнесены к расчёту молекулярно-механическим методом.

В местах разрыва связей между SE- и MM-частями связи в белка поместим т. н. «Link atoms». Для вычисления полной энергии молекулы или комплекса будем использовать схему с вычитанием: E = ESE(SE) + EMM(SE+MM) − EMM(SE). Зафиксируем атомные ядра лиганда и белка в положениях, полученных на предыдущем этапе.
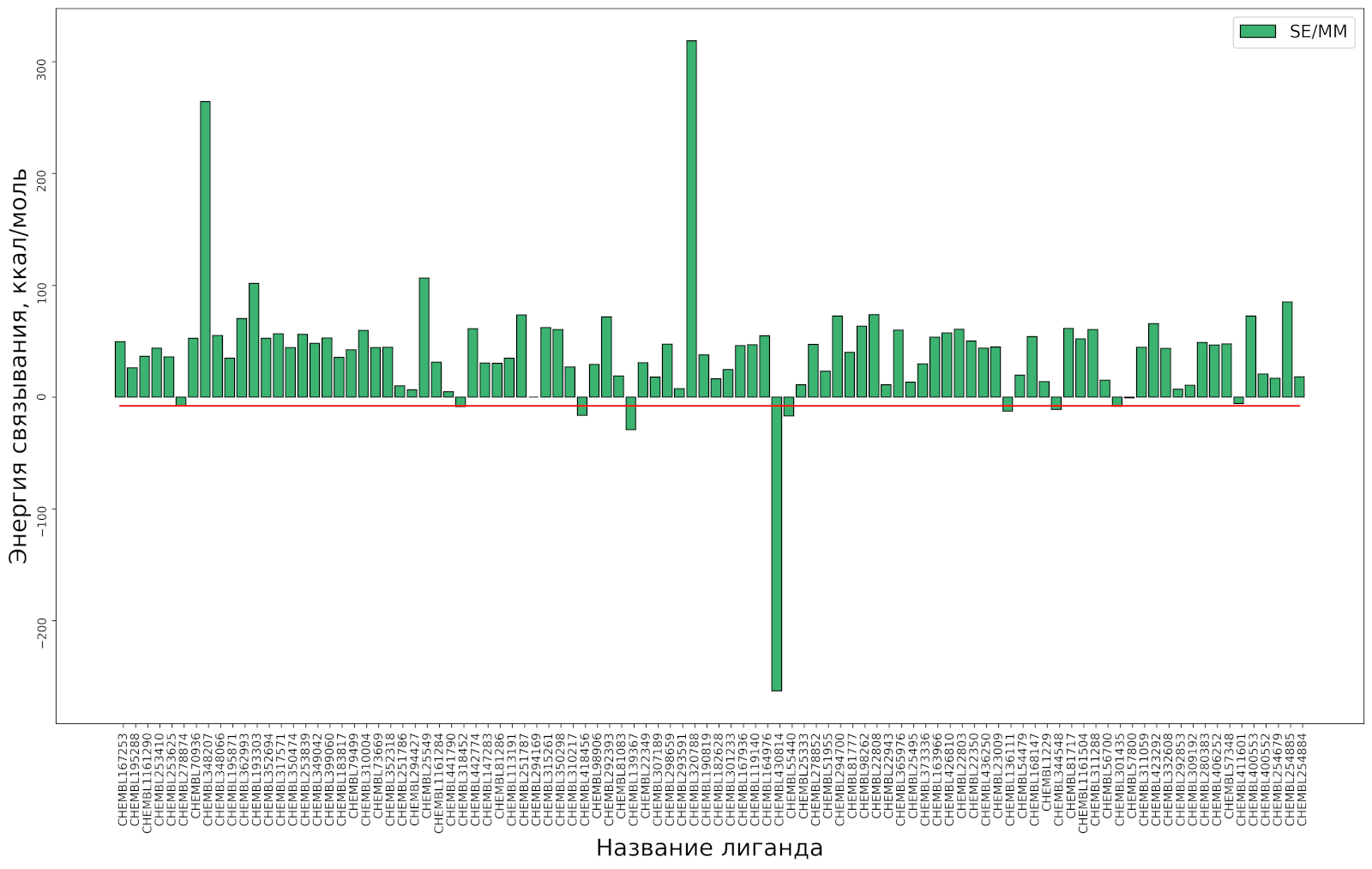
По результатам сделанного расчёта определилось связывание для 8 лигандов, неустойчивое связывание для 2 и отталкивание для 88. Повторим расчёт, используя не гибридную, а полуэмпирическую оценку энергии связывания (PM7) с использованием модели водной среды COSMO, продолжая фиксировать атомные ядра лиганда и белка в прежних положениях.

По результатам сделанного расчёта определилось связывание для 18 лигандов, неустойчивое связывание для 6 и отталкивание для 74. Однако заметим, что при этом использовались позы, оптимальные с т. з. эмпирической оценочной функции, которая, вообще говоря, не имеет соответствия с Ebind, рассчитанной по модели SE. Повторим расчёт, включив оптимизацию геометрии лиганда, используя в качестве начального приближения ранее зафиксированное положение.
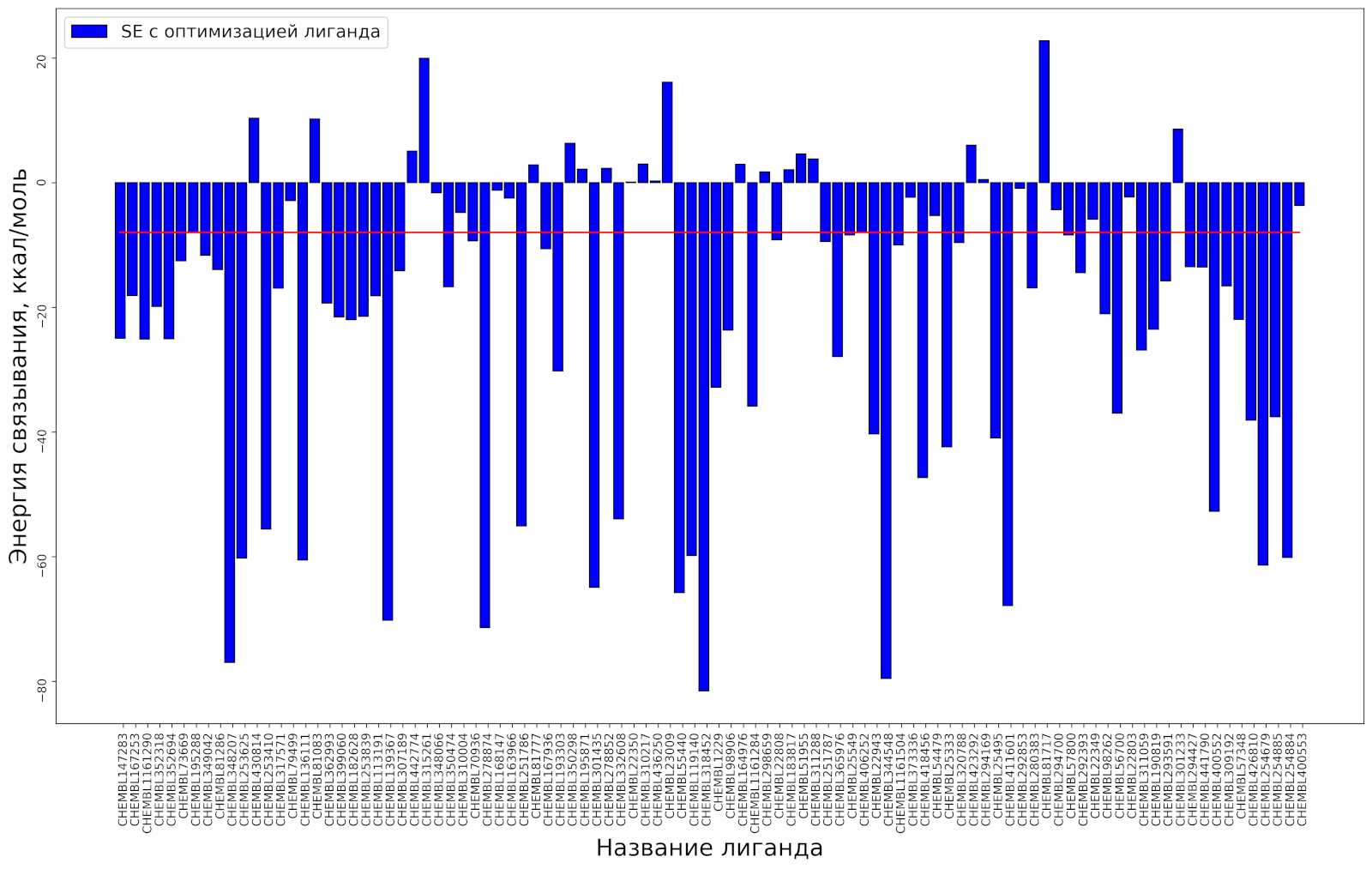
По результатам сделанного расчёта определилось связывание для 64 лигандов, неустойчивое связывание для 13 и отталкивание для 21.
Вывод: расчёт энергии связывания лигандов с белком по полуэмпирической модели (PM7), основанный на геометрии белка, полученной после оптимизации по модели силового поля (Amber) подтвердил связывание 65% лигандов и выдал ложноотрицательный результат для 35%, тогда как при расчёте эмпирическим методом (Vinardo) соотношение было 0/100.
Заметим, что проведённый глобальный поиск может выдавать произвольное количество поз, отранжированных по эмпирической оценочной функции; для данного расчёта бралась 1 лучшая поза для каждого из лигандов, однако при более тщательном подходе можно проводить оптимизацию по полуэмпирической модели, используя N лучших поз (например, N = 10).
Виртуальный скрининг
Рассмотрим вновь бромодомен PCAF и проведём виртуальный скрининг подмножества базы данных ChEMBL. Выбрав из базы данных соединения, относящиеся к классу «малых молекул» (small molecules) с молекулярным весом, находящимся в диапазоне от 100 до 200, получим подмножество из 34715 молекул, среди которых находится и вышеупомянутый ацетилированный лизин (NP2).
В ChEMBL содержатся лишь структуры молекул без координат атомных ядер. Для дальнейшей работы потребуются начальные приближения координат атомных ядер лигандов, которые можно вычислить с помощью метода фрагментов. После удаления соединений, для которых данный метод не дал результата, а также соединений, имеющих структурную несовместимость с моделью силового поля, используемой для лигандов, в списке останется около 33400 соединений.
Для каждого лиганда была с помощью глобального поиска определена оптимальная поза по оценочной функции Vinardo. Для полученных поз было рассчитано значение гибридной оценки энергии связывания (SE/MM) без оптимизации геометрии, т. е. по формуле Ebind = Eligand+PCAF − Eligand − EPCAF, где координаты атомных ядер во всех слагаемых соответствуют паре «лиганд-белок», оптимальной по оценочной функции Vinardo.

На втором этапе расчёта из списка лигандов, отранжированного по оценке энергии связывания SE/MM были отобраны 8340 лигандов (лучшие 25%). Для них было проведено вычисление энергии связывания с белком полуэмпирическим методом PM7 без оптимизации геометрии. Для 41 лиганда при вычислении энергии комплекса «лиганд-белок» метод самосогласованного поля не сошёлся; из оставшихся 8299 лигандов были выбраны 1245 (15%) с лучшей гибридной оценкой энергии связывания. На графике не показаны случаи образования ковалентной связи между белком и лигандом.
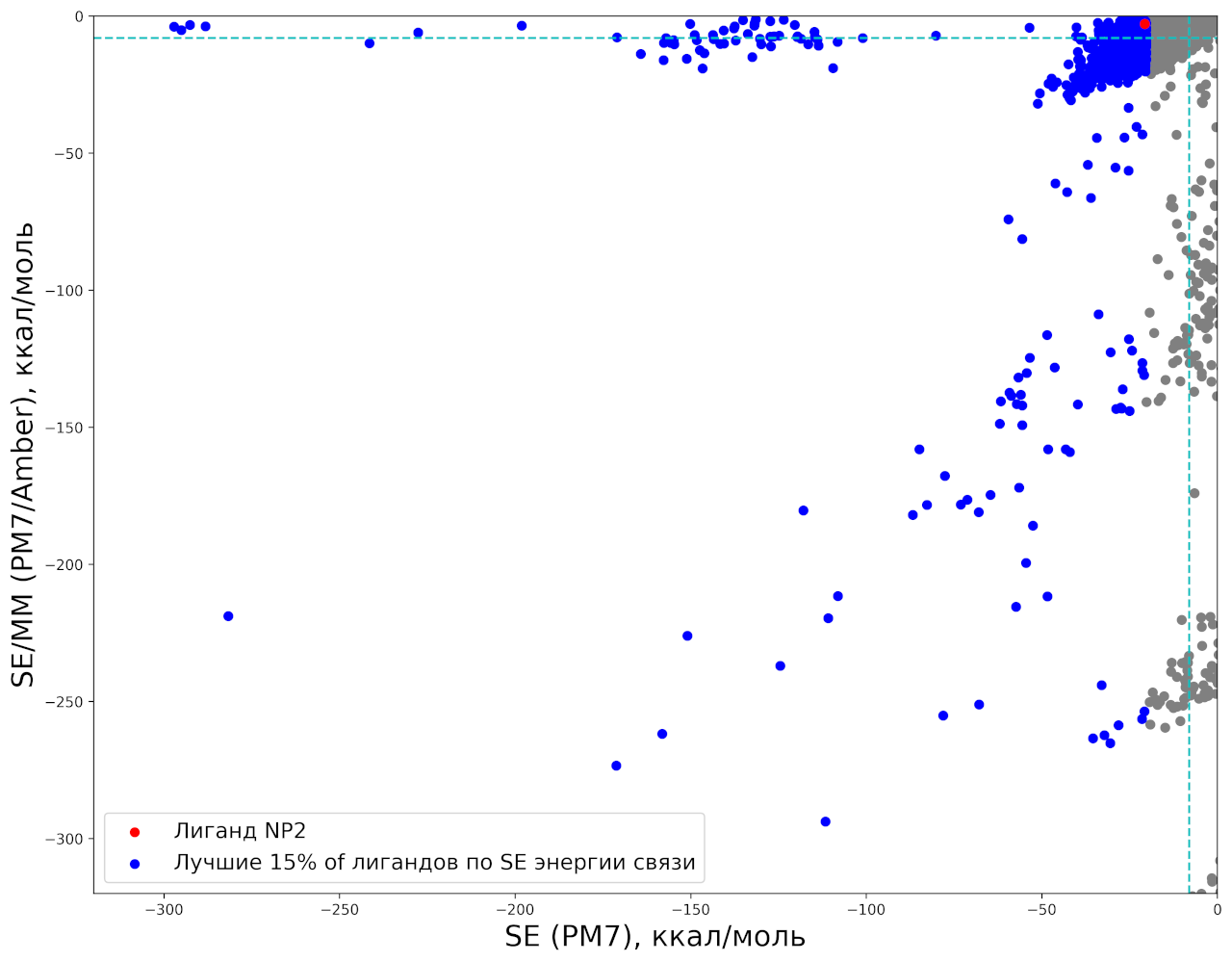
На третьем этапе для оставшихся 1245 лигандов было проведено вычисление энергии связывания методом PM7 с оптимизацией положений атомных ядер лиганда. Эта процедура закончилась успешно для 1200 лигандов; в остальных 45 не была достигнута сходимость процедуры оптимизации геометрии при расчёте энергии комплекса.

В ряде случаев оценка энергии связывания после оптимизации оказалась слабее, чем была без оптимизации; это обусловлено более точной оценкой энергии лиганда Eligand.
Вывод: в ходе виртуального скрининга были отобраны менее 3.5% от исходного множества соединений, выбранного из базы данных ChEMBL. Лиганд NP2, связывание которого подтверждено экспериментально, прошёл все этапы скрининга и присутствует в окончательном множестве.
Наиболее сильные оценки энергии связывания получили следующие 10 лигандов:
| Лиганд | Оценка энергии связывания |
|---|---|
| CHEMBL3277790 | 319.65 ккал/моль |
| CHEMBL373728 | 299.81 ккал/моль |
| CHEMBL3601593 | 282.58 ккал/моль |
| CHEMBL3601590 | 280.25 ккал/моль |
| CHEMBL3601582 | 278.68 ккал/моль |
| CHEMBL3601596 | 272.54 ккал/моль |
| CHEMBL3309713 | 224.57 ккал/моль |
| CHEMBL4069780 | 202.80 ккал/моль |
| CHEMBL1401401 | 200.85 ккал/моль |
| CHEMBL507116 | 198.23 ккал/моль |
Контакты
ООО «Энтрофорс», ИНН 7814680630
Электронная почта: info@entroforce.ru
Адрес: 199106 Санкт-Петербург, 16-я линия В.О., д. 7, пом. 6401